
To determine the concentration of copper in treated mine waste water samples by atomic absorption spectrometry, we can prepare a series of aqueous solutions containing a pure copper salt to calibrate the spectrometer and then use the resulting calibration graph in the determination of the copper in the test samples.
This approach is valid only if a pure aqueous solution of copper and a waste water sample containing the same concentration of copper give the similar absorbance values. In other words, by doing so we are assuming that there is no reduction or enhancement of the copper absorbance signal by other constituents present in the test sample. In many areas of analysis, this assumption is not always true. Matrix effect can have a significant influence to the final answer, even with methods such as plasma spectrometry (say ICP-AES) which is widely known for being relatively free from interferences.
There can be so-called proportional effects as these effects are normally proportional to the analyte signal, resulting in a change of the slope of the calibration curve.
One way to overcome this is to prepare the calibration standards in a matrix that is similar to the test sample but free of the targeted analyte, by adding known amounts of a copper salt to it in this discussion. However, in practice, this matrix matching approach is not practical. It will not eliminate matrix effects that differ in magnitude from one sample to another, and it may not be possible even to obtain a sample of the copper mine waste water matrix that contains no such analyte.
So, a better solution to this problem is that all the analytical measurements, including the establishment of the calibration graph, must in some way to be performed using the sample itself. Hence, the method of standard additions is proposed. It is widely practiced in atomic-absorption and emission spectrometry, and has also been applied in electrochemical analysis and many other areas.
This method of standard additions suggests to take six or more equal volumes of the sample solution, ‘spike’ them individually with known and different amounts of the analyte, and dilute all to the same volume. The instrument signals are then determined for all these standard solutions and the results are plotted as a linear calibration graph which has the signals plotted on the y-axis with its x-axis graduated in terms of the amounts of analyte added (either as an absolute weight or as a concentration).
When this linear regression is extrapolated to the point on the x-axis at which y = 0, we get a negative intercept on the x-axis which corresponds to the amount of the analyte in the test sample. So, when the linear calibration equation is expressed in the form of y = a + bx, where a is the y-intercept when x = 0, and b, the slope or gradient of the linear curve, simple geometry shows that the expected amount of the analyte in the test sample, xE, is given by a/b in absolute term, which is the ratio of the intercept and the slope of the regression line.
Since both a and b are subject to error, the calculated concentration is clearly subject to error as well. However, as this concentration is not predicted from a single measured value of y, the formula for the standard deviation, sxE of the sample analyte from the extrapolated x-value, xE is as follows:
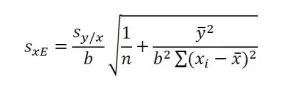
where sy/x is the standard error of y on x, and n, the number of points plotted on the regression. The standard error of y on x, sy/x of the linear regression is given by equation:
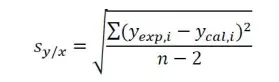
where yexp,i is the instrument signal value observed for standard concentration xi, and ycal,i, the ‘fitted’ yi-value calculated from the linear regression equation for xi value. This equation has made some important assumptions that the y-values obtained have a normal (Gaussian) error distribution and that the magnitude of the random errors in the y-values is independent of the analyte concentration (i.e. x-values).
Subsequently, we need to determine the confidence limits for xE as xE + t(n-2).sxE at alpha (a) error of 0.05 or 95% confidence. Increasing the value of n surely improves the precision of the estimated concentration. In general, at least six points should be used in a standard-additions experiment.
You may have noticed that the above equation differs slightly from the expression that is familiar in evaluating the standard deviation sx of an x-value given a known or measured y-value from a linear regression:
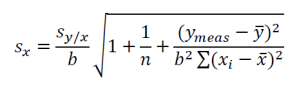
A worked example
The copper content in a sample of treated mining waste water was determined by FAAS with the method of standard additions. The following results were obtained:
Added Cu salt in moles/L 0.000, 0.0003, 0.006, 0.009, 0.012, 0.015
Absorbance recorded 0.312, 0.430, 0.584, 0.718, 0.838, 0.994
Let’s determine the concentration of copper level in the sample, and estimate the 95% confidence intervals for this concentration.
Apply the following equations for a, the y-intercept when x = 0, and b, the gradient or slope of the least-squares straight line expressed as y = a + bx:
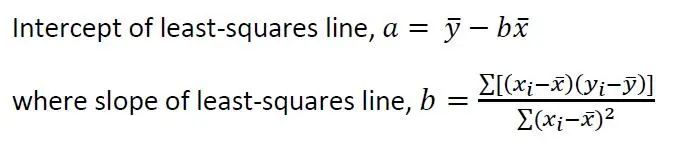
These two equations yielded a = 45.4095 and b = 0.3054. The ratio a/b gave the expected copper concentration in the test sample as 0.0067 moles Cu per liter.
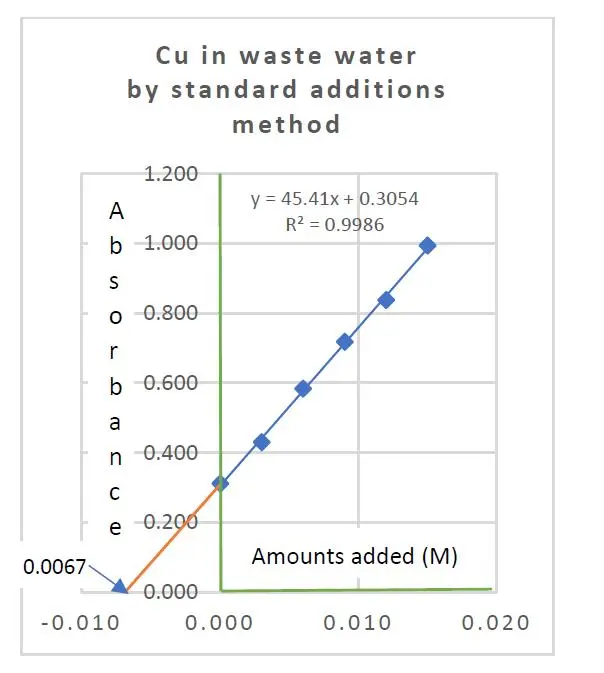
By further calculation, we have sy/x = 0.01048, sxE = 0.00028. Using Student’s t(6-2) critical value of 2.78, the confidence intervals are found to be 0.0067 M +/- 0.0008 M.
Although the method of standard additions is a good approach to cater for the common problem of matrix interference effects, it has certain disadvantages too:
You may also read our earlier article https://consultglp.com/2018/12/23/std-additions-in-instrumental-calibration/
ISO/IEC 17025:2017 accreditation standard requires laboratories to adopt standard test methods after verification of their performance, or in-house/non-standard methods after full validation. The main difference between verification and validation is the approaches to make sure the test methods adopted are fit for intended purpose.
Method verification is usually carried on standardized or internationally recognized methods which have been duly studied for their suitability. So, the laboratory needs only to show its technical competence in that it can meet the repeatability and reproducibility criteria laid down by the standard method concerned. Method validation, on the other hand, has to be conducted with many statistical parameters to confirm the suitability of the test method used.
Irrespective to either verification or validation, we must satisfy ourselves that the test methods adopted are precise and accurate. As we never know the true or native value of an analyte in a given sample sent for analysis, how can we be sure that the results presented to our customer are accurate or correct? How do we have confidence that the test method used in our laboratory is reliable enough for its purpose?
Routinely you may have carried out duplicates or triplicates in the analysis, but by doing so, you are actually studying the precision of the method based on the spread of test results in these replicated analyses. To know the accuracy of the method, you need to carry out analysis on some kind of samples with known or assigned value of the analyte to see if the recovery data are acceptable statistically. You may use a certified reference material (CRM) for this purpose.
ISO defines certified reference material (CRM) as a reference material characterized by a metrologically valid procedure for one or more specified properties, accompanied by a certificate that provides the value of the specified property, its associated uncertainty, and a statement of metrological traceability, while reference material (RM) is a material, sufficiently homogeneous and stable with respect to one or more specified properties, which has been established to be fit for its intended use in a measurement process.
Hence, one of its important usage in method validation is to assess the trueness (bias) of a method, although with careful planning of experiments, other useful information such as method precision can also be collected at the same time.
To know the accuracy of a test method is to monitor its biasness and recovery. Ideal samples in which analyte levels are well characterized, e.g.: matrix CRMs, are necessary. This is because pure analyte standards do not really test the method in the same way that matrix-based samples do. However, matrix CRMs may not be always available in the markets. If not available, then a reference material prepared in-house is our next best option.
In the absence of suitable reference materials, it is also possible to carry out recovery on spiked samples where known amounts of analyte are added to so-called ‘blank’ samples. However, the analyte in this case tends to be bound less closely in spiked samples than in real samples for analysis, and consequent recoveries tend to be over-estimated.
To understand the bias associated with an analytical method, we need first of all discuss measurement errors, which include random error and systematic error which lead us to bias (trueness).
We notice that repeated laboratory analyses always generate different results. This is due to many uncontrollable random effects during the experimentation. They can be assessed through replicate testing. However, experimental work is invariably subject to possible systematic effects too. A method can be considered ‘validated’ only if any systematic effects are duly studied and confirmed.
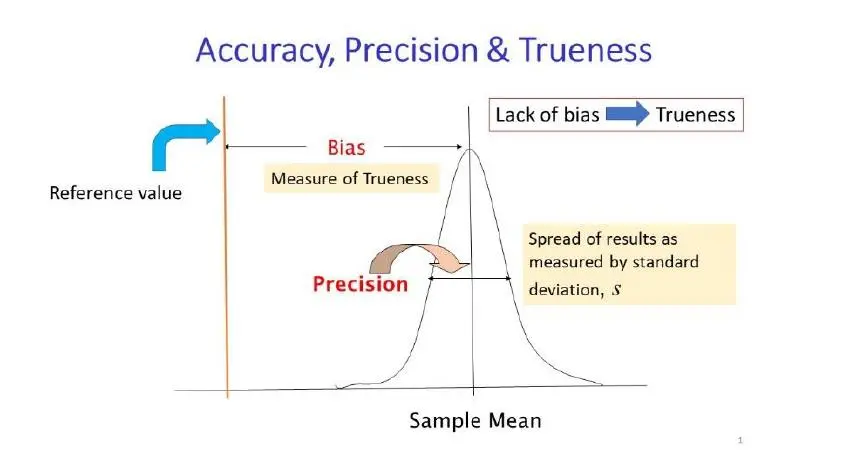
It is important to point out that under the current ISO definitions, accuracy is a property of a result and comprises both bias and precision, whilst the trueness is the closeness of agreement between the average value obtained from a large set of test results and an accepted reference value. ISO further notes that the measure of trueness is normally expressed in terms of bias.
The following figure gives an illustration of analytical bias and its relationship with precision of replicate analysis.
From the definitions of bias given, we know that:
It therefore follows that tests to measure bias need:
Bias can be expressed in one of the two ways:
The difference between a test result and its certified reference value does not tell us much about result bias. To know any significance between the difference, we have to carry out a series of replicate experiments and apply statistical treatment on the test data collected.
When conducting a bias study comparing the certified value for a reference material with the results obtained with the particular test method, we carry out a Student’s t -test statistic to interpret the results. We apply the mean value and its standard deviation of n replicates of the experiments in the following equation:
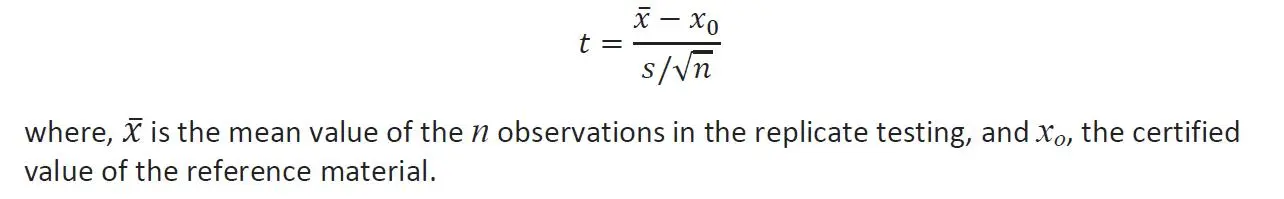
If t-value is greater than the t -critical value at alpha a error of 0.05, the bias is statistically significant with 95% confidence.
Bias information obtained during method development and validation is primarily intended to initiate any further method development and study. If a significant effect is found, action is normally required to reduce it to insignificance level. Typically, further study and corrective actions are to be taken to identify the source(s) of the error and bring it under control. Corrective actions might involve, for example, some changes to the test procedure or additional training.
However, it is quite unusual to correct an entire analytical method just for the sake of observed bias. If minor changes to the test protocols are not able to improve the accuracy of the results, we may resort to do a correction for recovery. If R is the average recovery noted in the experiments, a recovery correction factor of 1/R can be applied to the test results in order to bring them back to a 100% recovery level. However, there is no current consensus on such correction for recovery.
The Harmonized IUPAC Guidelines for the Use of Recovery Information in Analytical Measurement have recognized a rationale for either standardizing on an uncorrected method or adopting a corrected method, depending on the end use. Its recommendation is: It is of over-riding importance that all data, when reported, should (a) be clearly identified as to whether or not a recovery correction has been applied and (b) if a recovery correction has been applied, the amount of the correction and the method by which it was derived should be included with the report. This will promote direct comparability of data sets. Correction functions should be established on the basis of appropriate statistical considerations, documented, archived and available to the client.
Whilst attending an Eurachem Scientific Workshop on June 14-15, 2018 at Dublin of Ireland, the workshop organizer arranged the Workshop Banquet at the renowned Guinness St. James Gat Brewery, whose one of the employees was William Sealy Gosset, a chemist cum statistician.
Gosset was interested in analyzing quality data obtained small sample size in his routine work on quality control of raw materials, as he noticed that it was neither practical nor economical in analyzing hundreds of data.
At that time, making statistical inferences from small sample-sized data to their population was unthinkable. The general accepted idea was that if you were to have a large sample size, say well over 30 observations, you could use the Gaussian’s normal distribution to describe your data.
In 1906, Gosset was sent to Karl Pearson’s laboratory at the University College London on sabbatical. Pearson then was one of the well known scientific figures of his time, who was later credited with establishing the field of statistics.
At the laboratory, Gosset discovered the “Student’s t-distribution”, which is an important pillar of modern statistics to use small sample-sized data to infer what we could expect from the population out there. It is the origin of the concept of “statistical significance testing”.
Why didn’t Gosset name the distribution as Gosset’s instead of Student’s?
It is interesting to note that it was because his employer, Guinness objected to his proposal to publish the findings as it did not want the competitors to know their gained advantage in using this unique procedure to select the best varieties of barley and hops for their popular beer in a way that no other business could do.
So finally Gosset published his article on Pearson’s journal Biometrika in 1908 under the pseudonym “Student”, leading to the famous “Student’s t-distribution”.
In statistics and probability studies, the t-distribution is a probability distribution in dealing with a normally distributed population whilst the sample size is not large. It uses sample standard deviation (s) to estimate the population standard deviation (s) which is unknown. For small samples, the confidence limits of the population mean are given by:
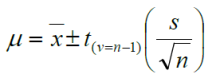
As the story goes, Gosset’s published paper was then mostly ignored by the statistical researchers until a young mathematician called R.A, Fisher discovered its importance and popularized it, particularly in estimating the random chance for considering a result “significant”.
Today, the t-distribution is routinely used as t- statistic tests for checking results for significance bias from true value, or for comparing measurements two sets of results and their means, and is also important for calculating confidence intervals.
This t-distribution is symmetric and resembles the normal distribution except for rather stronger “tails” due to more spread out because of the extra variability in smaller sample size.
