
A linear regression line showing linear relationship between independent variables (x’s) such as concentrations of working standards and dependable variables (y’s) such as instrumental signals, is represented by equation y = a + bx where a is the y-intercept when x = 0, and b, the slope or gradient of the line. The slope of the line becomes y/x when the straight line does pass through the origin (0,0) of the graph where the intercept is zero. The questions are: when do you allow the linear regression line to pass through the origin? Why don’t you allow the intercept float naturally based on the best fit data? How can you justify this decision?
In theory, you would use a zero-intercept model if you knew that the model line had to go through zero. Most calculation software of spectrophotometers produces an equation of y = bx, assuming the line passes through the origin. In my opinion, this might be true only when the reference cell is housed with reagent blank instead of a pure solvent or distilled water blank for background correction in a calibration process. However, we must also bear in mind that all instrument measurements have inherited analytical errors as well.
One of the approaches to evaluate if the y-intercept, a, is statistically significant is to conduct a hypothesis testing involving a Student’s t-test. This is illustrated in an example below.
Another approach is to evaluate any significant difference between the standard deviation of the slope for y = a + bx and that of the slope for y = bx when a = 0 by a F-test.
In a study on the determination of calcium oxide in a magnesite material, Hazel and Eglog in an Analytical Chemistry article reported the following results with their alcohol method developed:

The graph below shows the linear relationship between the Mg.CaO taken and found experimentally with equationy = -0.2281 + 0.99476x for 10 sets of data points.

The following equations were applied to calculate the various statistical parameters:

Thus, by calculations, we have a = -0.2281; b = 0.9948; the standard error of y on x, sy/x= 0.2067, and the standard deviation of y-intercept, sa = 0.1378.
Let’s conduct a hypothesis testing with null hypothesis Ho and alternate hypothesis, H1:
Ho : Intercept a equals to zero
H1 : Intercept a does not equal to zero
The Student’s t– test of y-intercept,

The critical t-value for 10 minus 2 or 8 degrees of freedom with alpha error of 0.05 (two-tailed) = 2.306
Conclusion: As 1.655 < 2.306, Ho is not rejected with 95% confidence, indicating that the calculated a-value was not significantly different from zero. In other words, there is insufficient evidence to claim that the intercept differs from zero more than can be accounted for by the analytical errors. Hence, this linear regression can be allowed to pass through the origin.
Most analytical methods require to determine the content of a constituent (analyte) in a given sample in terms of concentration as expressed as percentage by calculating ratio of the amount of the analyte and the weight of sample taken for analysis, multiplying by 100. For trace levels, we may calculate and expressed in terms of mg/kg, mg/L or even ug/L and pg/L, depending on how low the level is.
If w1, w2, …, wn are the weights of a series of samples and y1,y2, …, yn the corresponding measurements made on these samples, we usually calculate the ratios:

The average of these ratios is taken to represent the data collated. But we must remember that all measurements of y have a constant random error or bias, say, a. Hence, the ratios then become

We would expect the effect of a constant error in the measurement of y on the value of the ratio depends upon the magnitudes of w and y. We can avoid or conceal this disturbance in the ratio value, r, by making all the samples approximately the same weight taken for analysis. If the samples do cover an appreciable range of weights, the variation among the several values of the ratio not only reflects the inevitable random errors in y and w but is also dependent upon the weight taken from the sample.
Indeed, this is one of the important criteria in a method development process.
Let’s assume we take several weights (0.5g, 1.0g, 2.5g, 5.0g, 7.5g and 10.0g) of a certified reference fertilizer material containing 15.0% (m/m) K2O content for analysis. The absolute amounts of K2O present in these test samples are thus 0.075g, 0.15g, 0.375g, 0.75g, 1.125g and 1.50g, respectively. Upon analysis, the following results were obtained:

Figure 1 below shows the relationship between several paired values of the certified amount (x) and the analyzed value (y) graphically:

It is obvious that if there is no constant error in the analytical process, the various points should tend to lie closely along a line passing through the origin. But if there is a constant error or bias which is unavoidable, all the points are displaced either upwards or downwards the same amount. When the line intercepts the y-axis, this is the point corresponding to the ‘blank’ (i.e. zero analyte). Hence, the slope (or gradient) of the line, which shows the change in y for a unit change in x, is independent of the presence of a bias and has an advantage over the ratio, r.
So, to take this advantage, we should consider using the slope of line as an alternative means of combining the results of several determinations, provided the line remains linear over a reasonable range. This is because if all samples have the same weight as in this discussion, no information is forthcoming about the slope and we would not be able to detect the presence of a constant error.
Hence, we also see the advantage of linear regression in instrument calibration process where we plot the instrument signals against various concentrations of working standards. By so doing, we are able to estimate the errors of the slope & the y-intercept of the calibration curve, as well as the concentrations of sample solutions obtained from the calibration.
To start with, we apply the Ordinary Least Squares (OLS) formulae for fitting a straight line to a series of points on the graph. The line to be plotted must be at ‘equal distance’ among all the points where the sum of their ‘distances’, with positive and negative signs is zero. In here, the distance means the difference between the experimental y-value and the calculated y-value calculated from the linear equation. So, when we square these differences (i.e. deviations), the sum is a minimum or least positive figure.
It must be emphasized that an important but valid assumption on such regression line has been made, that is the recorded values on x-axis, sample weights in this case, are known exactly without error. For instrument calibration situation, it is assumed that the various working standard concentrations on the x-axis are of negligible error. It is indeed sufficient that the errors in x are small compared with the experimental errors in y (which have all the procedure steps as a source of random variation), and that the x’s cover an adequate range.

A simple computer program such as Microsoft Excel and R language will perform all these calculations, but most scientific calculators will not be adequate.
(to be continued in Part 2)
In instrumental analysis, there must be a measurement model, an equation that relates the amounts to be measured to the instrument response such as absorbance, transmittance, peak areas, peak heights, potential current, etc. From this model, we can then derive the calibration equation.
It is our usual practice to perform the experiment in such as a way as to fix influence standard concentration of the measurand and the instrument response in a simple linear relationship, i.e.,
y = a + bx ………. [1]
where
y is the indication of the instrument (i.e., the instrument response),
x is the independent variable (i.e., mostly for our purpose, the concentration of the measurand)
and,
a and b are the coefficients of the model, known as the intercept and slope (or gradient) of the curve, respectively.
Therefore, for a number of xi values, we will have the corresponding instrument responses, yi. We then fit the above model of equation to the data.
As usual, any particular instrumental measurement of yi will be subject to measurement error (ei), that is,
yi = a + bxi + ei …….. [2]
To get this linear model, we have to find a line that is best fit for the data points that we have obtained experimentally. We use the ordinary least square (OLS) approach, which chooses the model parameters that minimize the residual sum of squares (RSS) of the predicted y values versus the actual or experimental y values. The residual (or sometimes called error), in this case, means the difference between the predicted yi value derived from the above equation and the experimental yi value.
So, if the linear equation model is correct, the sum of all the differences from all the points (x, y) on the plot should be arithmetically equal to zero.
It must be stressed however, that for the sake of the above statement to be true, we make an important assumption, i.e., the uncertainty of the independent variable, xi, is very much less than in the instrument response, hence, only one error term ei in yi is considered due to this uncertainty which is sufficiently small to be neglected. Such assumption is indeed valid for our laboratory analytical purposes and the estimation process of measurement error is then very much simplified.
What is another important assumption made in this OLS method?
It is that the data are known to be homoscedastic, which means that the errors in y are assumed to be independent of the concentration. In other words, the variance of y remains constant and does not change for each xi value or for a range of x values. This also means that all the points have equal weight when the slope and intercept of the line are calculated. The following plot illustrates this important point.

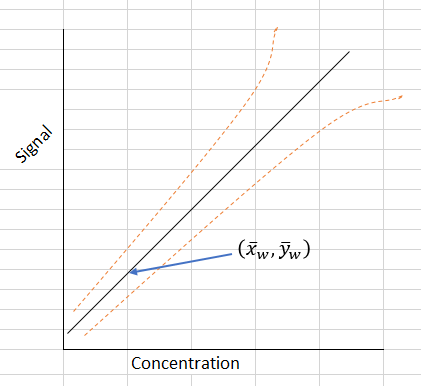
However, in many of our chemical analysis, this assumption is not likely to be valid. In fact, many data are heteroscedastic, i.e. the standard deviation of the y-values increases with the concentration of the analyte, rather than having the constant value of variation at all concentrations. In other words, the errors that are approximately proportional to the analyte concentration. In fact, we find their relative standard deviations which are standard deviations divided by the mean values are roughly constant. The following plot illustrates this particular scenario.

In this case, the weighted regression method is to be applied. The regression line must be calculated to give additional weight to those points where the errors are smallest, i.e. it is important for the calculated line to pass close to such points than to pass close to the points representing higher concentrations with the largest errors.
This is achieved by giving each point a weighting inversely proportional to the corresponding y-direction variance, si2. Without going through details of its calculations which can be quite tedious and complex as compared with those of the unweighted ones, , it is suffice to say that in our case of instrumental calibration which normally sees the experimental points fit a straight line very well, we would find the slope (b) and y-intercept (a) of the weighted line are remarkably similar to those of the unweighted line, and the results of the two approaches give very similar values for the concentrations of samples within the linearity of the calibration line.
So, does it mean that one the face of it, the weighted regression calculations have little value to us?
The answer is a No.
In addition to providing results very similar to those obtained from the simpler unweighted regression method, we find values in getting more realistic results on the estimation of the errors or confidence limits of those sample concentrations under study. It can be shown by calculations that we will have narrower confidence limits at low concentrations in the weighted regression and its confidence limit increases with increasing instrumental signals, such as absorbance. A general form of the confidence limits for a concentration determined using a weighted regression line is show in the sketch below:
These observations emphasize the particular importance of using weighted regression when the results of interest include those at low concentrations. Similarly, detection limits may be more realistically assessed using the intercept and standard deviation obtained from a weighted regression graph.
When there is a significant difference between the working calibration standard solutions for instrumental analysis and the sample matrix, its matrix background interference whilst using the calibration curve prepared by ‘pure’ standard solutions cannot be overlooked. Using the standard additions method for instrumental calibration is a good option. Read on …..

Suppose that we determined the amount of uranium contents in 14 stream water samples by a well-established laboratory method and a newly-developed hand-held rapid field method…..
A linear regression approach to bias between methods – Part II
Linear regression is used to establish a relationship between two variables. In analytical chemistry, linear regression is commonly used in the construction of calibration curve for analytical instruments in, for example, gas and liquid chromatographic and many other spectrophotometric analyses….
A linear regression approach to bias between methods – Part I
Generally speaking, linear regression is used to establish or confirm a relationship between two variables. In analytical chemistry, it is commonly used in the construction of calibration functions required for techniques such as GC, HPLC, AAS, UV-Visible spectrometry, etc., where a linear relationship is expected between the instrument response (dependent variable) and the concentration of the analyte of interest.
The word ‘dependent variable’ is used for the instrument response because the value of the response is dependent on the value of concentration. The dependent variable is conventionally plotted on the y-axis of the graph (scatter plot) and the known analyte concentration (independent variable) on x-axis, to see whether a relationship exists between these two variables.
In chemical analysis, a confirmation of such relationship between these two variables is essential and this can be establish in terms of an equation. The other aspects of the calibration can then be proceeded.
The general equation which describes a fitted straight line can be written as:
y = a + bx
where b is the gradient of the line and a, its intercept with the y-axis. The least-squares linear regression method is normally used to establish the values of a and b. The ‘best fit’ line obtained from the squares linear regression is the line which minimizes the sum of the squared differences between the observed (or experimental) and line-fitted values for y.
The signed difference between an observed value (y) and the fitted value (ŷ) is known as a residual. The most common form of regression is of y on x. This comes with an important assumption, i.e. the x values are known exactly without uncertainty and the only error occurs in the measurement of y.
Two mistakes are so common in routine application of linear regression that it is worth describing them so that they can be well avoided:
Some instrument software allows a regression to be forced through zero (for example, by specifying removal of the intercept or ticking a “Constant is ‘zero’ option”).
This is valid only with good evidence to support its use, for example, if it has been previously shown that y-the intercept is not significant after statistical analysis. Otherwise, interpolated values at the ends of the calibration range will be incorrect. It can be very serious near zero.
Sometimes it is argued that the point (x = 0, y = 0) should be included in the regression, usually on the grounds that y = 0 is an expected response at x = 0. This is actually a bad practice and not allowed at all. It seems that we simply cook up the data
Adding an arbitrary point at (0,0) will cause the fitted line to be more closer to (0,0), making the line fit the data more poorly near zero and also making it more likely that a real non-zero intercept will go undetected (because the calculated y-intercept will be smaller).
The only circumstance in which a point (0,0) can be validly be added to the regression data set is when a standard at zero concentration has been included and the observed response is either zero or is too small to detect and can reasonably be interpreted as zero.
